Informazioni per l’utilizzatore Inductos
ALLEGATO I
RIASSUNTO DELLE CARATT ER^TICHe DEL PRODOTTO
1. DENOMINAZIONE DEL MEDICINALE
InductOs 12 mg kit per impianto.
2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA
Un flaconcino contiene 12 mg di dibotermina alfa. Dopo ricostituzione InductOs contiene dibotermina alfa alla concentrazione di 1,5 mg/ml.
Dibotermina alfa (proteina-2 ossea morfogenica ricombinante di origine umana; rhBMP-2) e una proteina umana prodotta da una linea cellulare ovarica di criceto cinese (CHO).
Per l’elenco completo degli eccipienti consultare il paragrafo 6.1.
3. FORMA FARMACEUTICA
Kit per impianto.
Il Kit e composto da una polvere bianca per soluzione, da un sol vente lim pido e incolore e da una matrice bianca.
4. INFORMAZIONI CLINICHE
4.1 Indicazioni terapeutiche
InductOs e indicato per la fusione spinale anteriore lombare a livello singolo (L4-S1) in sostituzione del trapianto osseo autogeno in adulti con disturbo degenerativo del disco che hanno ricevuto almeno 6 mesi di trattamento non operativo per questa condizione.
InductOs e indicato per il trat.ame nto di f atture traumatiche della tibia nell’adulto, in aggiunta al consueto trattamento di riduzion' de’K fratture esposte e di fissazione di chiodi endomidollari non alesati.
Vedere paragrafo 5.1.
4.2 Posologia e modo di somministrazione
InductOs deve essere usato da un chirurgo specializzato nel settore.
PG_’olo'ia
Le indicazioni per la preparazione di ciascun kit devono essere seguite esattamente, usando la dose appropriata di InductOs per l’indicazione voluta.
In assenza completa di esperienza, l’uso ripetuto del medicinale non e raccomandato.
Intervento di fusione spinale anteriore lombare
Il numero di porzioni di InductOs necessari e determinate dalla grandezza del Dispositivo LT-CAGE Lumbar Tapered Fusion utilizzato. Usando la tabella qui sotto, identificare il numero di porzioni di InductOs da 2,5 X 5 cm richiesti dalla grandezza del Dispositivo LT-CAGE Lumbar Tapered Fusion.
|
Grandezza del dispositivo LT-CAGE Lumbar Tapered Fusion (diametro principale x lunghezza) |
Numero di porzioni di InductOs di 2.5 x 5 cm per il dispositivo LT-CAGE Lumbar Tapered Fusion |
|
14 mm x 20 mm |
1 |
|
14 mm x 23 mm |
1 |
|
16 mm x 20 mm |
1 |
|
16 mm x 23 mm |
2 |
|
16 mm x 26 mm |
2 |
|
18 mm x 23 mm |
2 |
|
18 mm x 26 mm |
2 |
Fratture traumatické della tibia
II numero di kit di InductOs e il volume di InductOs al momento dell’impianto dipendono dall’anatomia della frattura e dall’abilita di chiudere la ferita s'nza stip are o '«■ iprimere eccessivamente il prodotto. In genere e sufficiente il contenuto di un kit p m trattare una frattura. Il dosaggio massimo di InductOs e limitato a due kit.
Popolazione pediatrica
La sicurezza e l’efficacia di InductOs nei bambini sotto i 18 anni non sono state stabilite.
Non ci sono dati disponibili.
Modo di somministrazione
Il prodotto ricostituito si somministra mediant’ impianto.
Durante la preparazione, la manipolazione e l’impianto del prodotto, evitare un’eccessiva perdita di liquido da InductOs. Non strizzare.
Per le istruzioni sulla ricostituzione del medicinale prima della somministrazione, vedere paragrafo 6.6.
Intervento di _fusione spinale anteriore lombare
InductOs non ' eve ess-re usato da solo per questa indicazione, ma deve essere utilizzato con il Dispositivo LT-Cage Lumbar Tapered Fusion.
Una eventual^ omusione nel seguire le istruzioni di preparazione del prodotto per InductOs potrebbe compromettere la sua sicurezza ed efficacia. Attenzione e cautela devono essere utilizzate per p -e/emm un ->empimento eccessivo del dispositivo e/o dello spazio intervertebrale (vedere paragrafo 4.4).
Pre-impianto
Tagliare la matrice bagnata di InductOs in 6 porzioni uguali (all’incirca 2,5 X 5 cm). Durante il taglio e nel maneggiare la matrice, evitare un’eccessiva perdita di fluido da InductOs. Non strizzare.
Usando le pinze chirurgiche per evitare un eccessivo strizzamento, con attenzione arrotolare il numero richiesto di porzioni di InductOs per ogni dispositivo LT-CAGE ed inserire ogni rotolo nel corrispettivo Dispositivo LT-CAGE Lumbar Tapered Fusion, come descritto nella figura sottostante.
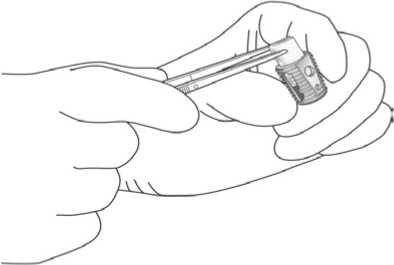
Impianto
II materiále discale e le porzioni cartilaginose delle placche vertebrali terminali devono essere rimo sse preservando le porzioni corticali delle placche vertebrali terminali, secondo la prassi stam ard.
Per le istruzioni su come impiantare il Dispositivo LT-CAGE Lumbar Tapered Fusion fare riferimento al foglietto illustrativo del dispositivo LT-CAGE.
Post-impianto
Una volta impiantati InductOs e il dispositivo LT-CAGE, non irrigare la zona dello spazio intervertebrale.
Se e necessario un drenaggio chirurgico, posizionare il drenaggio lontano dal sito dell’impianto o, preferibilmente, ad uno strato superficiale al sito dell’impianto.
Istruzioni per l ’uso nelle _ fratture traumatiche della tibia
Pre-impianto
• Prima di procedere all’impianto di InductOs occorre ridurre completamente la frattura, stabilizzarla ed indurre emostasi.
• InductOs non fornisce stabilita mec,am a e non deve essere usato per riempire spazi in presenza di forze di compressione.
• Piegare o tagliare InductOs come necessario prima di procedere all’impianto. Se l’intervento chirurgico richiede che solo una porzione del prodotto sia utilizzata, prima preparare l’intero prodotto InductOs (veu’re paragraf , 6.6), tagliare il prodotto nel formato desiderato e gettare le porzioni non utilizzate.
Impianto
Si procede all’imp.aito d h^^Os dopo aver completato il trattamento standard della frattura e della ferita, vale a dire quando si procede alla chiusura dei tessuti molli. Per quanto possibile, con InductOs va ricoperta la sípei^a' accessibile della frattura (linee di frattura e difetti). Disporre InductOs in modo che ada a colmare l’area della frattura e sia bene a contatto con i frammenti distali e prossimali maggiori. Per ottenere l’effetto desiderato non e necessario sovrapporre il contenuto di piú kit.
InductOs essere collocato in uno spazio vuoto (poco compresso), piegato, arrotolato o avvolto a seconda della geometria della frattura.
Post-impianto
Dopo aver proceduto all’impianto di InductOs, non irrigare la ferita.
Se e necessario un drenaggio chirurgico, collocare il drenaggio in un punto lontano dal sito dell’impianto o, preferibilmente, ad uno strato piú superficiale rispetto al sito dell’impianto.
Al fine di ottenere la massima potenza di efficacia, e importante fare in modo che InductOs dopo il suo impianto risulti completamente coperto da tessuti molli.
4.3 Controindicazioni
InductOs e controindicato nei pazienti con:
• Ipersensibilitá al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
• Immaturitá scheletrica.
• Qualsiasi tumore maligno in atto o in pazienti sottoposti a terapia antitumorale.
• Un focolaio di infezione nel punto di intervento.
• Persistenza della sindrome da compartimento o residui neurovascolari della sindrome stessa.
• Fratture patologiche quali quelle osservate (ma non limitate ad esse) nel morbo di Paget o delle ossa con metastasi.
4.4 Avvertenze speciali e precauzioni d’impiego
Una eventuale omissione nel seguire le istruzioni di preparazione del prodotto per InductOs potrebbe compromettere la sua sicurezza ed efficacia. Attenzione e cautela devono essere utilizzate per prevenire un eccessivo riempimento del dispositivo e/o dello spazio intervertebrale.
Intervento di chirurgia spinale cervicale
Edema localizzato associato all’uso di InductOs e stato riportato in pazienti soť oposti a 'hnurgia spinale cervicale. L’insorgenza dell’edema e stata tardiva e generalmente si e ven^cata nella prima settimana dopo l’operazione. In alcuni casi l’edema e stato abbastanza grave da provocare una compromissione delle vie respiratorie. La sicurezza e l’efficacia dell’uso di InductOs in chirurgia spinale cervicale non e stata stabilita e InductOs non deve essere utilizzato in tale situazione.
Tumori maligní
InductOs non deve essere utilizzato in pazienti con presenza in anamnesi o sospetto clinico di tumore maligno nel sito di applicazione (vedere paragrafo 4.3).
Formazione di raccolte di liquidi
Formazione di raccolte di liquidi (pseudocisti, edema localizzato, essudato nella sede dell’impianto), a volte incapsulate, in alcuni casi provocanť campxssi 'ne dei nervi e dolore sono state riportate in pazienti sottoposti a chirurgia spinale associata con l’ uso di InductOs. Molte di queste segnalazioni si sono verificate quando InductOs veniv usato :n s chemi terapeutici/dispositivi non approvati o in modo non coerente alle istruzioni per l’uso. Un intervento clinico (aspirazione e/o rimozione chirurgica) potrebbe essere richiesto se i sintomi persistono (vedere paragrafo 4.8).
Riassorbimento osseo
InductOs puo causare inizialmente il riassorbimento dell’osso trabecolare circostante. Pertanto, in assenza di dati clinici, il prodotto non deve essere usato per una applicazione diretta sull’osso trabecolare quando il LranJtorio riassorbimento puo creare rischi di fragilitá dell’osso. Quando InductOs e stcM ualizz ato con il dispositivo LT-CAGE (vedere paragrafo 4.2 ) in studi clinici per la fusione spinale anteriore lombare, la frequenza e la gravitá di riassorbimento dell’osso, come evidenziato dalle iperdiafanie radiologiche e/o dalla migrazione del dispositivo, erano simili a quelle osservate nei pazienti trattati con trapianto osseo autogeno.
Oss'fj'azione e. eropica
L’uso di InductOs puo causare ossificazioni eterotopiche nei tessuti circostanti che possono causare complicazioni. Sono state osservate ossificazioni al sito dell’impianto e una formazione ectopica dell’osso.
Compressione dei nervi
E’ stata riportata compressione dei nervi associata alla formazione ectopica di osso e all’utilizzo di InductOs (vedere paragrafo 4.8). Potrebbe rendersi necessario un ulteriore intervento chirurgico.
Migrazione del dispositivo
Dopo l’utilizzo di InductOs in interventi chirurgici di fusione spinale puo verificarsi migrazione del dispositivo, che puo richiedere revisione chirurgica (vedere paragrafo 4.8).
Risposta immunitaria
Sia la dibotermina alfa che il collagene bovino di Tipo I hanno provocato reazioni immunitarie nel paziente.
Anticorpi anti-dibotermina alfa: negli studi di fusione spinale anteriore lombare, lo 0,7 % dei pazienti che hanno ricevuto InductOs hanno sviluppato anticorpi contro lo 0,8% dei pazienti che hanno ricevuto il trapianto autogeno dell’osso. Negli studi sulla frattura traumatica della tibia il 4,4% dei pazienti che ricevevano InductOs hanno sviluppato anticorpi, contro lo 0,6% osservato nel gruppo di controllo.
Anticorpi anti-collagene bovino di Tipo I: negli studi di fusione spinale anteriore lombare, il 19% dei pazienti che hanno ricevuto InductOs hanno sviluppato anticorpi al collagene bovino di Tipo I contro il 13% dei pazienti che hanno ricevuto il trapianto autogeno dell’osso. Negli studi sulla frattura traumatica della tibia il 15,7% dei pazienti che ricevevano InductOs hanno sviluppato anticorpi, contro l’ 11,8% nel gruppo di controllo. In entrambe le due indicazioni, nessun paziente risultato positivo agli anticorpi anti-bovino del collagene di Tipo I ha sviluppato antn >rpi al collagene umano di Tipo I.
Sebbene negli studi condotti nell’uomo non siano stati osservati manifestazioni cliniche o effetti collaterali, la possibilitá che si sviluppino anticorpi neutralizzano o reazioni di ipersensibilitá non e esclusa. Per i pazienti che in precedenza hanno ricevuto college p^r via sistemica deve essere fatta una oculata valutazione del rischio/beneficio (vedere paragrafo 4.3). Nei casi in cui si sospetta che il manifestarsi di reazioni avverse sia legato a precedenti problemi di origine immunologica, occorre valutare la possibilitá che il prodotto possa provocare una reazione immunitaria.
Popolazioni speciali
La sicurezza e l’efficacia dell’uso di InductOs in pazienti con malattie autoimmuni accertate non e stata stabilita. Queste malattie autoimmuni includono artrite reumatoide, lupus eritematoso sistemico, sclerodermia, sindrome di Sjogren e dermatomiosite/polimiosite.
La sicurezza e l’efficacia ď Ind ctOs ion s^no state dimostrate in pazienti con disturbi metabolici ossei.
Non sono stati condotti stud’ sui pazienti con ridotta funzionalitá epatica o renale. Per tutti questi gruppi di pazienti e opport mo che il medico valuti attentamente benefici e rischi del paziente specifico prima di aťizzare ^ductOs. Si raccomanda uno stretto monitoraggio del paziente per rilevare eventuali reazioni avverse e confermare il buon esito del trattamento.
Eccipienti
Questo medicinale contiene meno di 1 mmol (23 mg) di sodio per dose massima (2 kit), cioe e praticamente “senza sodio”.
Avvertenze speciali e precauzioni d’impiego specifiche per la fusione spinale anteriore lombare
La sicurezza e l’efficacia di InductOs non e stata stabilita nelle seguenti condizioni:
• usato con impianti spinali diversi dal dispositivo LT-CAGE
• impiantato in siti diversi dal livello lombare inferiore L4-S1
• usato in tecniche chirurgiche diverse da approcci anteriori aperti o laparascopici anteriori
Quando il disturbo degenerativo del disco e stato trattato con una procedura di fusione intersomatica posteriore lombare con gabbiette cilindriche filettate e dibotermina alfa, in alcuni casi e stata osservata un’ossificazione posteriore dell’osso.
Avvertenze speciali e precauzioni d’impiego specifiche per le fratture traumatiche della tibia
InductOs e concepito per l’uso in pazienti con le seguenti caratteristiche:
• riduzione e stabilizzazione della frattura ,adeguate, per garantire la stabilita meccanica.
• stato neurovascolare adeguato (ad es. assenza della sindrome da compartimento, basso rischio di amputazione).
• emostasi adeguata (che consenta di disporre di una superficie per l’impianto relativamente asciutta).
• assenza di esteso difetto di riparazione delle ossa lunghe, in cui puo verificarsi una notevole compressione dei tessuti molli.
L’impianto deve essere posizionato sulla frattura sotto una adeguata visione e con la massima cura (vedere paragrafo 4.2).
Informazioni relative all’efficacia sulla frattura della tibia provengono soltanto da studi clinici controllati nei quali sono state trattate fratture esposte della tibia con inserimento di chiodi endomidollari nell’osso (vedere paragrafo 5.1). In uno studio clinico in cui il canale endomidollare era alesato a “chatter” corticale, e stato osservato un aumento dell’incidenz- di >mezion' nel gruppo trattato con InductOs rispetto al gruppo di controllo sottoposto al trattamento standard (vedere paragrafo 4.8). Non si raccomanda l’utilizzo di InductOs con chiodi alesati nella riparazione delle fratture esposte della tibia.
InductOs non fornisce stabilita meccanica e non deve essere usato per riempire spazi vuoti in presenza di forze di compressione. Il trattamento della frattura di o sa n ngh~ , dei tessuti molli deve basarsi sulle terapie standard, compreso il controllo dell’infezione.
4.5 Interazioni con altri medicinali ed altre forme d’interazione
Non sono stati eseguiti studi di inte.azicne.
Poiché dibotermina alfa e una proteina e non iei. riscontrata nella circolazione corporea, e un candidato improbabile per un’interazione farmacocinetica farmaco-farmaco.
Informazioni da studi clinici suUe fratture traumatiche della tibia indicano che l’uso di InductOs in pazienti sottoposti a terapia con glucocorticoidi non e stato associato alla comparsa di alcuna reazione avversa evidente. In studi p' ^clinici la somministrazione contemporanea di glucocorticoidi ha ridotto la ricostruzione dell’osso (misurata come percentuale nei confronti di un gruppo controllo), ma l’efficacia di InductO s non e s.ata alterata.
In studi clinici sulla frattura traumatica della tibia in cui i pazienti trattati con InductOs ricevevano per un periodo di 14 giorni consecutivi FANS in associazione, si sono manifestati effetti indesiderati medi o moc eraui ccreF i alla guarigione (ad esempio con il drenaggio della ferita) in numero maggiore risp^tto a quNlo osservato nei pazienti trattati con InductOs ma che non assumevano FANS. Sebbene il risultato finale per il paziente non sia stato modificato non si puo escludere un’interazione tra FANS e InductOs.
4.6 Fertilita, gravidanza e allattamento
Gravidanza e donne potenzialmente fertili
Non vi sono dati adeguati provenienti dall’utilizzo di dibotermina alfa nelle donne in stato di gravidanza.
Gli studi su animali hanno dimostrato tossicita riproduttiva (vedere paragrafo 5.3).
Il rischio potenziale per le donne e sconosciuto.
Sono stati eseguiti studi su gli animali che non possono escludere effetti degli anticorpi anti-dibotermina alfa sullo sviluppo embrio-fetale (vedere paragrafo 5.3). Poiché non si conoscono i rischi per il feto associati al potenziale sviluppo di anticorpi neutralizzanti per la dibotermina alfa, InductOs non deve essere usato durante la gravidanza a meno che non sia chiaramente necessario (vedere paragrafo 4.4). Le donne in eta fertile devono essere avvisate di utilizzare un efficace metodo contraccettivo per almeno 12 mesi dopo il trattamento.
Allattamento
Non e noto se dibotermina alfa e escreta nel latte umano. L’escrezione di dibotermina alfa non e stata studiata negli animali. L’allattamento non e raccomandato durante il trattamento con InductOs.
Fertilita
Negli studi sull’animale non e stato rilevato alcun impatto sulla fertilita. Non sono disponibili dati clinici; il potenziale rischio nell'uomo non e noto.
4.7 Effetti sulla capacita di guidare veicoli e sull’uso di macchinari
Considerando che InductOs non ha effetti sistemici, e probabile c he Indu'tOs non alteri o alteri in modo trascurabile la capacita di guidare veicoli o di usare medona'i.
4.8 Effetti indesiderati
Sintesi del profilo di sicurezza
Le piú comuni reazioni averse correlate all’uso di InductOs sono state nevralgia negli interventi di chirurgia spinale e infezione localizzata negli interventi di riparazione delle fratture della tibia. La reazione avversa piú grave e edema lo 'aEzza, o negli interventi di chirurgia spinale cervicale. Sull’incidenza delle reazioni avverse associate a InductOs non hanno influito sesso, eta o razza.
Tabella delle reazioni avverse
Negli studi clinici piú di 1600 pazienti hanno ricevuto InductOs. I pazienti con frattura delle ossa lunghe trattati con Ind-ctC s sono stati oltre 485. Negli studi di fusione spinale, quasi 900 pazienti sono stati trattati con InductOs. L’ultimo gruppo di pazienti ha preso parte agli studi sull'uso di InductOs per wi'azitni non ancora approvate nell’UE. Questi dati sono integrati con informazioni sull’uso di InductOs nella popolazione generale.
Nella tabella che segue e presentata la frequenza delle reazioni avverse osservate in pazienti esposti al t attame nto c on InductOs. La frequenza e indicata con i termini molto comune (> 1/10) o comune (> 1/100; < 1/10). Non sono state rilevate reazioni con frequenza definibile dagli aggettivi non comune (> 1/1.000; < 1/100), raro (> 1/10.000; < 1/1.000) o molto raro (< 1/10.000).
La frequenza delle reazioni avverse rilevate durante l’uso post-commercializzazione di InductOs non e nota poiché tali reazioni sono state riferite da una popolazione di numerosita incerta.
|
Classificazione per sistemi e organi |
Frequenza | ||
|
Molto comune |
Comune |
Non nota | |
|
Patologie sistemiche e condizioni relative |
Migrazione del |
Edema localizzato* | |
|
alla sede di somministrazione |
dispositivo1 * |
Pseudocisti* Essudato nella sede dell’impianto * | |
|
Patologie del sistema muscoloscheletrico e deltessuto connettivo |
Ossificazione extrascheletrica, calcificazione eterotopica postoperatoria, aumento della formazione di tessuto osseo (formazione ectopica di osso)1* Mal di schiena1 Disturbo osseo1 |
Osteolisi* Aumento dell’assorbimento osseo* | |
|
Patologie del sistema nervoso |
Nevralgia1 |
Radiculite1 | |
|
Infezioni e infestazioni |
Infezione localizzata2* |
1 Osservati durante l’uso in procedure di fusione spinale anteriore lombare
2 Osservata durante l’uso in interventi su fratture traumatiche della tu >'
* Ulteriori informazioni sono fornite di seguito
Descrizione di alcune reazioni avverse Rimodellamento osseo
Il rimodellamento osseo si verifica nell’ambito de’ mexanism■ di azione farmacologico di InductOs (vedere paragrafo 5.1). In questo processo avvengono sia il riassorbimento osseo sia la formazione di nuovo tessuto osseo. In alcune circostanze aueAi pioces°; possono portare a complicanze, quali compressione dei nervi (dovuta all? formation ’ di tessuto osseo o a calcificazione eterotopica) oppure migrazione del dispositivo (associata a riassorbimento osseo o osteolisi).
Infezione localizzata
Una infezione localizzata specifica dell’arto fratturato e stata molto comune (> 1/10) nei pazienti in uno studio clinico in cui il cana.’ eidoniidollare e stato alesato a “chatter” corticale. Una frequenza aumentata di infezione ’ s^ta osservata nel gruppo trattato con InductOs rispetto al gruppo di controllo sottoposto a1 trať am^nto standard (rispettivamente 19% versus 9%; vedere paragrafo 4.4). Per l’uso con chiodi non alesati, le incidenze stimate di infezione in uno studio sono state simili tra i gruppi di trattamento (rispettivamente 21% versus 23%).
Raccolta di fluidi (edema localizzato, pseudocisti, essudato nella sede dell ’impianto)
In considerazione dell’attivita angiogenica di InductOs puo verificarsi la formazione di raccolte di fluidi (pseudociti, edema localizzato, essudato nella sede dell’impianto), a volte incapsulati, che in alcuni cas ca^ano compressione dei nervi e/o dolore.
Un edema localizzato si osserva molto comunemente quando si utilizza InductOs in interventi di fusion' spinale della colonna cervicale. L’edema ha mostrato un’insorgenza ritardata e, in alcuni casi, e stato abbastanza grave da provocare una compromissione delle vie respiratorie (vedere paragrafo 4.4).
4.9 Sovradosaggio
L’uso di InductOs in pazienti sottoposti a chirurgia spinale cervicale in concentrazioni o quantitá maggiori di quelle raccomandate nel paragrafo 4.2 per le indicazioni approvate, e stato associato a casi di edema localizzato (vedere paragrafo 4.4).
Nel caso di pazienti riceventi concentrazioni o quantitá maggiori di quelle raccomandate, il trattamento deve essere di supporto.
5. PROPRIETÁ FARMACOLOGICHE
5.1 Proprieta farmacodinamiche
Gruppo farmacoterapeutico: farmaci per il trattamento delle malattie ossee, Proteina Ossea Morfogenetica; codice ATC: M05BC01
Dibotermina alfa e una proteina osteoinduttiva che stimola l'osteogenesi nel sito dell’impianto. Dibotermina alfa si lega ai recettori presenti sulla superficie delle cellule mesenchimali e n e promuove la differenziazione in cellule cartilaginee ed ossee. Con la degradazione della matrice le cellule differenziate formano l’osso trabecolare determinando al tempo stesso una evidente vascolarizzazione. L'osteogenesi si sviluppa partendo dalla parte esterna dell’im punto em~ d centro finché tutto l’impianto di InductOs e sostituito da osso trabecolare.
Il rimodellamento dell’osso trabecolare avviene secondo modalitá conform al’e mrze biomeccaniche esercitate sulla parte. L'applicazione di InductOs nell’osso trabecolare etermin a un riassorbimento transitorio dell’osso che circonda l’impianto, che in seguito viene sostituito da neoformazione ossea piú densa. La capacitá di InductOs di sostenere l’osso in fase ^ rimodellamento puo determinare l’integrazione biologica e biomeccanica della neoformazione o sma indotta da InductOs con quella dell’osso circostante. Valutazioni radiografiche, biomeccaniche e istologiche dell’osso indotto indicano che funziona biologicamente e biomeccanicamente come l’osso autologo. Inoltre studi preclinici hanno indicato che l’osso indotto da InductOs, se fratturato, si ripara in modo indistinguibile rispetto all’osso autologo.
Studi preclinici hanno suggerito che losteoge^si prodotta da InductOs e un processo autolimitante, capace cioe di costituire soltanto un volume len p"eciso di osso. Questa autolimitazione e dovuta probabilmente alla perdita di dibotermina alfa dal sito dell’impianto nonché dalla presenza di inibitori delle BMP nei tessuti circostanti. Inoltre numerosi studi preclinici indicano che vi e un meccanismo di feedback negativo a livello m detmlam ch e limita la osteoinduzione da parte delle BMP.
Studi di farmacologia clinica dim ostraho che la matrice da sola non e osteoinduttiva e che non e piú presente in biopsie effettuate a sole 16 settimane dalla data dell’impianto.
Informazioni farm aco dinami 'v e specifiche agli studi sulla fusione spinale anteriore lombare
L’efficacia e la úcumzz ' di InductOs sono state dimostrate in uno studio randomizzato, controllato, multicentrico, di non inferioritá su 279 pazienti di etá compresa tra i 19-78 anni, sottoposti a una procedura in aperto di fusione anteriore lombare. I pazienti ricevevano almeno 6 mesi di trattamento non operativ o prima di ricevere InductOs per la fusione spinale anteriore lombare. I pazienti venivano randomizzati a ricevere il dispositivo LT-CAGE Lumbar Tapered Fusion riempito con InductOs o con il trapianto autogeno dell’osso prelevato dalla cresta iliaca.
24 mesi dopo l’intervento, InductOs si e dimostrato statisticamente non inferiore al trapianto autogeno dell’osso. La percentuale di successo della fusione valutata radiologicamente era del 94,4% per InductOs contro l’88,9% (intervallo di confidenza della differenza:-1,53, 12,46; 95% test a due code) per il trapianto autogeno dell’osso. Per il dolore e l’invaliditá (punteggio Oswestry), la percentuale di successo era del 72,9% contro il 72,5 % (intervallo di confidenza della differenza:-11,2, 12,0; 95% test a due code). Un endpoint singolo, multi composito definito come successo globale era la prima variabile dello studio. Il successo globale consisteva delle seguenti valutazioni primarie di efficacia e di sicurezza:
1 Fusione dimostrata radiologicamente Doc umento reso disponibile da AIFA il 12/06/2014 10
2 Miglioramento del punteggio Oswestry per il dolore e l’invalidita
3 Mantenimento o miglioramento dello stato neurologico
4 Nessuno evento avverso classificato di grado 3 o di grado 4 valutato come associato all’impianto o alla procedura di impianto/chirurgica
5 Nessuna procedura aggiuntiva chirurgica eseguita e classificata come un “insuccesso”
24 mesi dopo l’operazione, il tasso di successo globale era del 57,5% per InductOs contro il 55,8% (intervallo di confidenza della differenza:-10, 72, 14, 01; 95% test a due code) per il trapianto autogeno dell’osso.
Uno studio ulteriore, non comparativo di 134 pazienti che hanno ricevuto trattamento di fusic^ intersomatica anteriore lombare tramite tecniche chirurgiche laparoscopiche, ha prodotto la stessa percentuale di successo rispettivamente di 92,9% per la fusione, 85,6% per dolore e invali ’ita e 90,3% per gli stati neurologici. Lo studio ha confermato l’applicabilita della fusione . pin. 'e ante^ore lombare utilizzante InductOs tramite tecniche chirurgiche di impianto laparoscopico.
Informazioni farmacodinamiche specifiche agli studi sulla frattura traumatica de.'a tu 'a
L’efficacia di InductOs era stata dimostrata in uno studio multinaz. onale, ra ^om^zato, controllato, in singolo cieco, condotto su 450 pazienti (di eta tra 18 e 87 anni; 81% maschi) .on fratture esposte della tibia che richiedevano una terapia chirurgica. I pazienti sono stati sottoposti (in una proporzione pari a 1:1:1) a una cura standard della frattura e della terapia dei tessuti molli (la cura standard includeva l’impiego di chiodi endomidollari), alla cura standard piú InductOs 0,75 mg/ml, o cura standard piú InductOs 1,5 mg/ml. I pazienti venivano seguiti per i successivi12 mesi dopo la chiusura dei tessuti molli.
Nello studio registrativo sulla frattura traumatica della tibia ha evidenziato che InductOs accresce le probabilita di guarigione. I pazienti trattati con .ndu^tOs alla concentrazione di 1,5 mg/ml presentavano una riduzione del 44% del rischio di insuccesso della terapia (necessita di un intervento secondario per favorire la guarigione) "isp etto ai p azienti che erano stati sottoposti alla cura standard (RR = 0,56; 95% CI = da 0,40 a 0,78). Questi risultati sono stati convalidati da controlli radiologici condotti senza essere a conoscenza del tipo di terapia effettuata. Il numero di interventi secondari e successivi era notevolmente ridotto nei pazienti trattati con InductOs, soprattutto in relazione a interventi piú invasivi come il tra pianto osseo e la sostituzione dei chiodi (P=0,0326).
La proporzione dei pazienti guariti dopo il trattamento con InductOs 1,5 mg/ml era significativamente piú alta a tutte le visite fatte dalla decima settimana fino ai 12 mesi dopo l’intervento, suggerendo una accelerazione della guarigione della frattura.
Il trattamento con InductOs 1,5 mg/ml si e rivelato efficace significativamente (se confrontato alla cura standard) in tutti i pazienti indipendentemente dalla loro storia di fumatori.
Gr wit' del'' fr ’tture: il trattamento con InductOs 1,5 mg/ml risultava estremamente efficace per tutte le c1. ,si di fratt re, comprese le fratture gravi Gustilo IIIB (riduzione del 52% del rischio di un intervento secondario rispetto ai soggetti sottoposti a cure standard).
Dopo 6 settimane dal trattamento la percentuale di pazienti con guarigione delle lesioni dei tessuti molli era significativamente superiore nel gruppo che aveva ricevuto InductOs 1,5 mg/ml rispetto al gruppo sottoposto a cure standard (83% rispetto al 65%; P = 0,0010). La percentuale di pazienti in cui e stato osservato il cedimento degli impianti (piegatura o rottura delle viti di ancoraggio) era significativamente piú basso nel gruppo trattato con InductOs 1,5 mg/ml rispetto al gruppo sottoposto a cure standard (11% rispetto al 22%; P = 0,0174).
5.2 Proprieta farmacocinetiche
InductOs e attivo nel sito dell’impianto. In due studi esplorativi sono stati prelevati campioni di siero prima e dopo l'intervento da alcuni pazienti che presentavano fratture delle ossa lunghe. Non e stata riscontrata la presenza di dibotermina alfa nel siero.
In studi condotti su animali (ratti) usando InductOs contenente dibotermina alfa radiomarcata, la durata media di permanenza nel sito dell’impianto era di 4-8 giorni. I picchi di dibotermina alfa in circolazione (0,1% della dose impiantata) si registrano entro 6 ore dall’impianto. Quando iniettato per via endovenosa, l'emivita terminale di dibotermina alfa e pari a 16 minuti nel ratto e 6,7 minuti nella scimmia “cynomolgus”. Da quanto sopra esposto si conclude, quindi, che dibotermina alfa viene rilasciato lentamente dalla matrice nel sito dell’impianto e viene invece rapidamente elimi ato una volta entrato nella circolazione sistemica.
5.3 Dati preclinici di sicurezza
Dati non clinici rivelano assenza di rischi per l’uomo sulla base di studi cor' enJona’ di farmacologia, di tossicitá acuta e di tossicitá per somministrazioni ripetute.
In studi di tossicitá riproduttiva nei ratti, dove la dibotermina alfa veniva somministrata per endovena per massimizzare l’esposizione sistemica, sono stati osservati un accresciuto peso fetale e un’aumentata ossificazione nel feto e non e stato possibile escludere un effetto correlato al trattamento. La rilevanza clinica di questi effetti e sconosciuta.
Gli anticorpi anti-dibotermina sono stati studiati in coniglie in stato di gravidanza dopo iper-immunizzazione con dibotermina alfa per indurre sperimentalme nte anticorpi anti-BMP-2.
In alcuni feti con pesi corporei diminuiti, vi era una di únuzione nell’ossificazione delle ossa frontali e parietali (4 feti su 151), considerata generalm ^/e rever ;bile, e non e stato possibile escudere gli effetti dovuti agli anticorpi. Non vi sono state altre alterazioni nella morfologia fetale esterna, viscerale o scheletrica. Altri studi su gli animali non indicano effetti dannosi diretti o indiretti sulla gravidanza, tossicitá materna, embrioletalitá, o fetotossicitá.
InductOs non e stato sottoposto a studi di carcinogenicitá in vivo. La dibotermina alfa ha mostrato effetti variabili su linee cellmari ťmorali umane in vitro. Nonostante i dati disponibili in vitro suggeriscano un basso potenzian di s+imolazione della crescita tumorale, l’uso di InductOs e controindicato in p^iei ti con tumo 5 maligni in atto o in pazienti sottoposti a terapia antitumorale (vedere paragrafo 4.3).
InductOs e stat o Audia to in un modello animale di impianto spinale canino. InductOs e stato impiantato direttamente sulla dura esposta a seguito di laminectomia. Benché siano stati osservati restringim* nto del neuioforamen e stenosi, non sono state riscontrate né la mineralizzazione della dura, né stenosi della spina dorsale, né deficit neurologici susseguenti al trattamento con InductOs. Il significato di questi dati per gli uomini e sconosciuto.
6. iNFORMAZIONI farmaceutiche
6.1 Elenco degli eccipienti
Polvere
Saccarosio
Glicina
Acido glutammico Sodio cloruro Polisorbato 80 Sodio idrossido.
Solvente
Acqua per preparazioni iniettabili.
Matrice
Collagene bovino Tipo I
6.2 Incompatibilita
Questo medicinale non deve essere mescolato ad altre sostanze medicinali ad eccezione di quelle citate nel paragrafo 6.6.
6.3 Periodo di validita
3 anni
6.4 Precauzioni particolari per la conservazione
Non conservare al di sopra dei 30°C. Non congelare.
Conservare nella confezione originale per proteggere il medicinale dalla luce.
6.5 Natura e contenuto del contenitore Ogni kit di InductOs contiene:
• 12 mg di polvere di dibotermina alfa sterile .n u flaconcmo da 20 ml (vetro Tipo I) chiuso con
tappo in gomma bromobutile, sigillato con una ghiera in alluminio e capsula in plastica.
• Solvente per ricostituzione in un fla^nc^o da 10 ml (vetro Tipo I ) chiuso con tappo in gomma bromobutile ,sigillato con una ghiera in alluminio e capsula in plastica .
• Una matrice sterile in una vasch 'tta m poli ■ nlcloruro (PVC) sigillata con una lamina di Tyvek.
• Due siringhe sterili monouso da 10 ml in polipropilene.
• Due aghi sterili (acciaio inossidabile).
6.6. Istruzioni particolari per lo smaltimento e altre indicazioni
InductOs viene prer ara^o imm>4iatamente prima dell’uso. La dibotermina alfa deve essere utilizzata soltanto con il solvente e la matrice forniti in dotazione nel kit di InductOs.
Una volta preparato InductOs contiene dibotermina alfa alla concentrazione di 1,5 mg/ml (12 mg per flaconcino). InductOs non deve essere usato a concentrazioni superiori a 1,5 mg/ml (vedere paragrafo 4.9).
Preparazione del prodotto
In campo non sterile
1. Usando una tecnica sterile, collocare nel campo sterile una siringa, un ago e la matrice nella sua confezione.
2. Disinfettare i tappi dei flaconcini di dibotermina alfa e del solvente.
3. Usando la siringa rimanente e l’ago dal kit, ricostituire il contenuto del flaconcino di dibotermina alfa con 8.4 ml di solvente. Iniettare lentamente il solvente nel flaconcino
contenente la dibotermina alfa liofilizzata. Ruotare delicatamente il flaconcino per facilitare la ricostituzione. Non agitare. Dopo l’uso eliminare la siringa e l’ago.
I
8.4 ml solvente
4. Disinfettare il tappo del flaconcino di dibotermina alfa ricostituito.
In campo sterile
5. Sollevando il lato superiore, aprire la confezione contenente la matrice e lasciare la matrice nel suo vassoio.
6. Utilizzando la tecnica del trasferimento asetfico e la sAng a e l’ago della fase 1, prelevare 8 ml di soluzione di dibotermina alfa ricostituita dal flaconcino del campo non sterile, sollevando il flaconcino capovolto per facilitare il prelievo.
7. Lasciando la matrice nel vassfio, distri’ Jre UNiFORMEMENTE la soluzione di dibotermina alfa sulla matrice seguendo lo schema indicato nella figura sottostante.
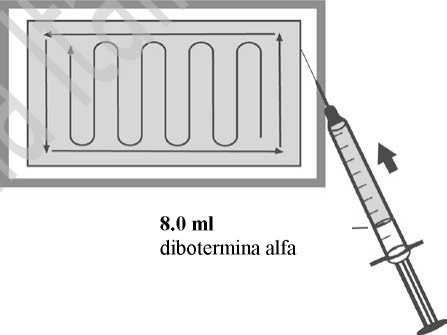
8. Aspettare ALMENO 15 minuti prima di utilizzare il prodotto InductOs cosi preparato. Il prodotto deve essere usato entro due ore dalla preparazione.
Per evitare di sovraccaricare la matrice, e importante ricostituire la dibotermina alfa e bagnare l’intera spugna come descritto in precedenza.
9. Seguire le istruzioni relative all’intervento chirurgico pianificato - fusione spinale anteriore lombare o riparazione della frattura traumatica della tibia.
Il prodotto non utilizzato ed i rifiuti derivati da tale medicinale devono essere smaltiti in conformitá ai requisiti di legge locali.
7. TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
Medtronic BioPharma B.V.
Earl Bakkenstraat 10 6422 PJ Heerlen Paesi Bassi
tel +31 (0) 45 566 8000 fax +31 (0) 45 566 8012
8. NUMERO(I) DELL’AUTORIZZAZIONE(DELLE AUTORIZZAZION i)
ALL’IMMISSIONE IN COMMERCIO
EU/1/02/226/001
9. DATA DELLA PRIMA AUTORIZZAZIONE/DEL RINNOVO DELL’AUTORIZZAZIONE
Data della prima autorizzazione: 9 settembre 2002 Data di ultimo rinnovo: 20 luglio 2012
10. DATA DI REVISIONE DEL TESTO
Informazioni piú dettagliate su questo medici, 'alt ^ono disponibili sul sito web della Agenzia europea dei medicinali http://www.ema.europa.eu/
A. PRODUTTORE(I) DEL(DEI) PRINCIPIO(i) A"TIVO(I) BIOLOGICO(I) E PRODUTTORE(I)RESPONSABILE(I) DEL RILASCIO DEI LOTTI
B. CONDIZIONI O LIMITAZIONI DI foRNITURA E DI UTILIZZO
C. ALTRE CONDIZIONI E REQ UIs HI DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMerCIO
D. CONDIZIONI O LIMITAZIONI PER QUANTO RIGUARDA L’USO SICURO ED EFficACE DEL MEDICINALE
A. PRODUTTORE(I) DEL PRINCIPIO(I) ATTIVO(I) BIOLOGICO(I) E PRODUTTORE(I) RESPONSABILE(I) DEL RILASCIO DEI LOTTI
Nome ed indirizzo del (dei) produttorc(i) del(dei) principio(i) attivo(i) biologico(i)
Wyeth BioPharma One Burtt Road Andover
Massachusetts 01810 USA
Nome ed indirizzo del produttore(i) responsabile del rilascio dei lotti
Medtronic BioPharma B.V.
Earl Bakkenstraat 10 6422 PJ Heerlen Paesi Bassi
B. CONDIZIONI O LIMITAZIONI DI FORNITURA E DI UT lIZZO
Medicinale soggetto a prescrizione medica speciale e limitativa (vedere allegato I: riassunto delle caratteristiche del prodotto, paragrafo 4.2).
C. ALTRE CONDIZIONI E REQUISITI DELL' AUTOrIZZAZIONE ALL'IMMISSIONE IN COMMERCIO
• Rapporti periodici di aggiornamento sulla sicurezza
Il titolare dell’autorizzazione all’ir missione ;n commercio deve fornire i rapporti periodici di aggiornamento sulla sicurezza per questo medicinale conformemente ai requisiti definiti nell’elenco delle date di riferimento per l’Unione europea (elenco EURD) di cui all’articolo 107 quater, par. 7) della direttiva 2010/84/CE e pubMiu'to sul portale web dei medicinali europei.
D. CONDIZION ' O LIMTAZIONI PER QUANTO RIGUARDA L’USO SICURO ED EFFICACE DEL MEDICINALE
• Piano di gestione del rischio (RMP)
Il titolare dell’autorizzazione all'immissione in commercio deve effettuare le attivita e gli interventi di farmacovigilanza richiesti e dettagliati nel RMP concordato e presentato nel modulo 1.8.2 dell’autorizzazione all'immissione in commercio e qualsiasi successivo aggiornamento concordato del RMP.
Il RMP aggiornato deve essere presentato:
• su richiesta dell’Agenzia europea per i medicinali;
• ogni volta che il sistema di gestione del rischio e modificato, in particolare a seguito del ricevimento di nuove informazioni che possano portare a un cambiamento significativo del profilo beneficio/rischio o al risultato del raggiungimento di un importante obiettivo (di farmacovigilanza o di minimizzazione del rischio).
Se la presentazione dello PSUR e del RMP aggiomato coincide, PSUR e RMP possono essere presentati allo stesso tempo.
ETICHETTATURA E FOGLIO ILLUSTRATIVO
Doc umento reso disponibile da AIFA il 12/06/2014 19